Teaching AI the Language of Life: Inside the Rise of Genomic Language Models
Why researchers began treating DNA like a language, why the analogy works surprisingly well, and where it breaks. A tour through genomic language models: tokenization, training, the context-length problem, and the new architectures built for biology.

🧬 Genomic AI is moving fast and it will likely affect far more people than just computational biologists.
With the rise of personalized and precision medicine, genomics is becoming a foundational discipline for healthcare, drug discovery, and biotechnology. Yet many of the AI models now being developed to understand genomes remain difficult to explain outside specialist circles.
I think this is a problem.
Genomic AI is reshaping medicine but its inner workings stay hidden from most. Let’s shed some light.
If genomic AI will shape how we diagnose diseases, design therapies, and understand biology, then the basic ideas behind these models should be understandable to more than just domain experts.
So here is a short attempt to explain one of the central ideas in the field:
Why researchers started treating DNA like a language and why that idea is both powerful and imperfect.
🧬 The language of life
Large Language Models (LLMs) like GPT work by learning patterns in sequences of text. They learn grammar, relationships between words, and how meaning changes depending on context.
Now consider DNA.
Instead of an alphabet of dozens of letters, DNA uses just four characters:
A, T, G, C.
And instead of forming sentences, these letters form long sequences encoding the instructions for life.
At first glance, the analogy becomes obvious:
DNA looks a lot like text.
This observation led researchers to ask an interesting question:
Could the same AI models that understand human language also learn the patterns hidden in genomic sequences?
This idea led to the development of Genomic Language Models (gLMs), models that apply language‑modeling techniques to biological sequences.
For simplicity, we will mainly focus on DNA sequences in this article. However, similar language‑model approaches are also used to study other biological sequences, including RNA and protein (amino acid) sequences.
✨ Why this idea works surprisingly well
In natural language, the meaning of a word depends on the surrounding words.
For example:
“The researcher studied the expression of a gene.”
“The artist showed great expression in her painting.”
The same word appears in both sentences, but its meaning changes depending on the context.
Something similar happens in genomics.
A short DNA motif might act as:
- a promoter: a DNA pattern located just before a gene that helps start the process of reading that gene
- an enhancer: a DNA pattern that can sit far away from a gene but increases how strongly that gene is activated
- a splice signal: a sequence pattern that tells the cell where to cut and reconnect RNA when building the final gene message
but only depending on the sequence context around it.
This is exactly the type of relationship that transformers are designed to learn.
Transformers process sequences using layers of attention heads, where each head learns to focus on different patterns within the input.
For example, one of the most commonly used models for local experimentation today is LLaMA‑3.1 8B.
It consists of 32 layers, and each layer contains 32 attention heads.
In natural language, different heads may capture:
- grammar
- syntax
- semantic relationships
When applied to DNA, those same mechanisms can learn:
- regulatory motifs
- transcription factor binding patterns
- genomic sequence structure.
Several genomic models apply transformer architectures directly to DNA sequences, including:
These models showed that large neural networks can learn meaningful biological signals directly from raw genomic data.
🧩 But DNA is not just another language
The analogy between language and genomes is powerful, but it quickly reaches its limits.
First, we cannot read or interpret genomic sequences directly.
Humans can look at a sentence and immediately understand its meaning. Even without formal training in linguistics, we intuitively grasp words, grammar, and context.
DNA is different. Unlike a sentence in human language, a string of nucleotides such as
GCTTACGATCGTACGTTAGC…
does not carry obvious meaning to us. We cannot directly interpret which parts encode genes, which regulate expression, how distant regions interact, or what functional role a sequence ultimately plays in the cell.
Second, the vocabulary is extremely small.
Human languages use tens of thousands of words.
DNA only has four nucleotides.
Third, DNA sequences can be interpreted in both directions.
Human language is usually read in a single direction. English, for example, is written and interpreted from left to right.
DNA, however, is double‑stranded. Genes and regulatory elements can appear on either strand of the molecule, meaning biologically meaningful patterns may occur in a sequence or in its reverse complement.
For example, the DNA sequence:
ATGC
has a reverse complement:
GCAT
A regulatory motif might appear in either orientation, meaning genomic models often need to recognize patterns that appear when a sequence is read from left to right or from right to left something language models trained on text rarely have to consider.
Fourth, genomes are not purely linear.
While we write DNA sequences as long strings of letters, inside the cell DNA folds into complex 3D structures. Regions that are far apart along the sequence can physically interact with each other.
A helpful analogy one of my professors once shared:
Inside the cell nucleus, DNA is packed into a structure called chromatin - the active working form of DNA. You can imagine it like a string of pearls, where the pearls represent genomic elements like genes along the DNA strand.
If you stretch the string out, two pearls might be very far apart.
But if you drop the string onto a desk, it wrinkles and folds. Suddenly pearls that were previously far apart along the string can end up sitting right next to each other.
Something similar happens in the genome. DNA folds and loops inside the nucleus, bringing distant regions of the sequence into close physical proximity.
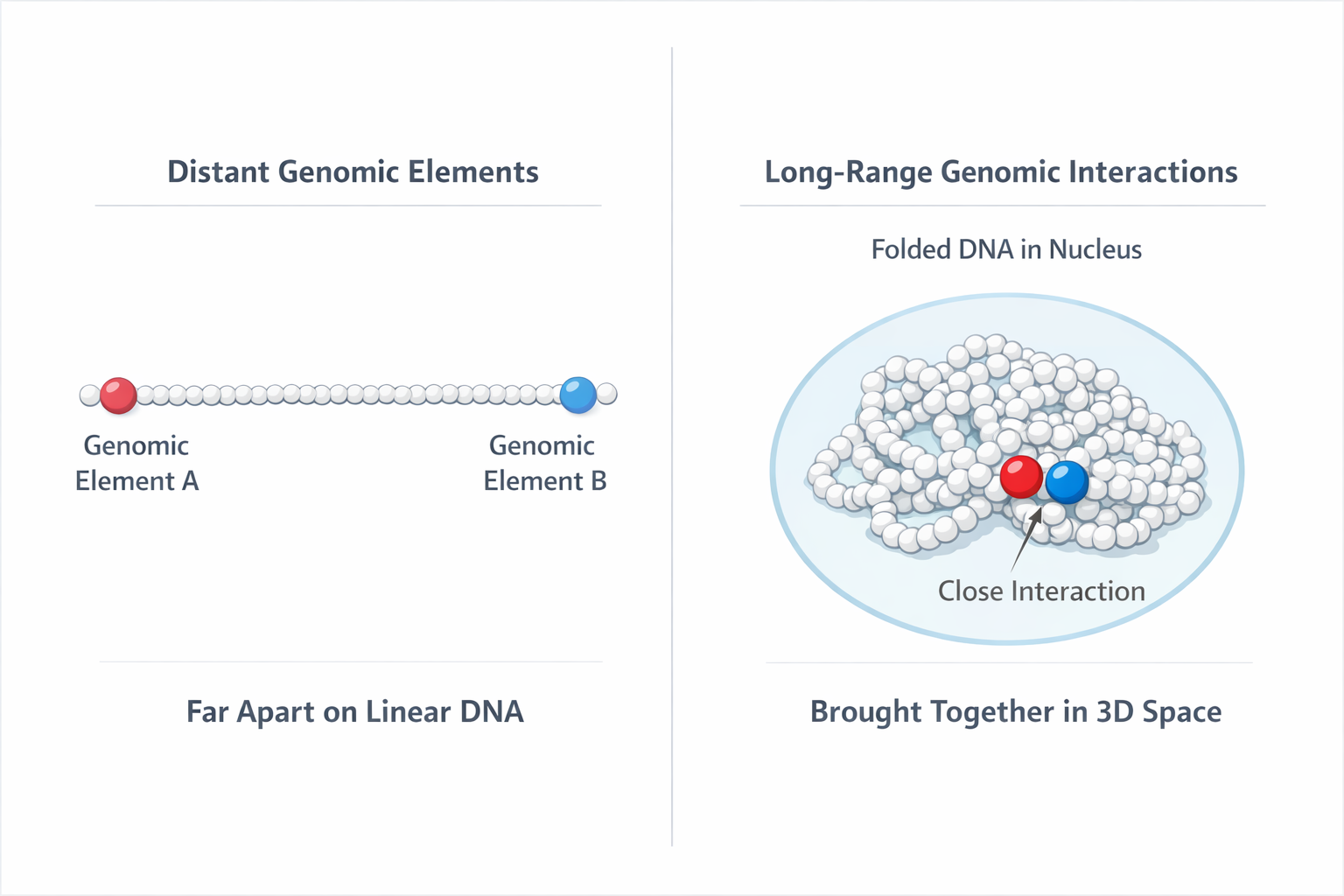
String-of-pearls analogy: chromatin folding brings distant genomic regions into close physical proximity
Sometimes regulatory elements located millions of base pairs away influence whether a gene is activated.
These long‑range interactions create a major challenge for AI models trying to learn patterns in genomic sequences.
📏 The context length challenge
For AI models trying to learn patterns in DNA, this creates a major challenge.
Transformers, the architecture behind most modern language models, rely on an attention mechanism that scales with O(n²) complexity.
This means every token in a sequence attends to every other token.
Modern language models can handle very long context windows, sometimes hundreds of thousands or even millions of tokens. However, in natural language most meaningful relationships occur within relatively short ranges. Words rarely need to interact with other words hundreds of pages away at high resolution.
So for sentences or documents this works quite well.
But genomes are different, and genomes are enormous.
Important regulatory interactions can occur hundreds of thousands or even millions of base pairs apart, meaning models must capture long‑range dependencies across the sequence, ideally while maintaining resolution at the level of individual nucleotides.
This makes scaling sequence models for genomics particularly challenging, both computationally and architecturally.
A practical example of this limitation can be seen in protein structure prediction models, which also rely on sequence modeling.
One interesting model is ESMFold, which is based on the ESM protein language model. These models treat amino‑acid sequences similarly to how language models treat text and can predict the 3D structure of proteins directly from their sequence.
ESMFold is remarkably fast compared to models like AlphaFold, especially for relatively small structures, because it does not rely on large evolutionary alignment databases. This allows structures to be predicted with very low latency.
However, the model still inherits a fundamental limitation from transformer architectures: the O(n²) scaling of attention.
What does that mean in practice?
If a protein has 800 amino acids, the model needs to consider roughly:
800² ≈ 640,000 pairwise interactions.
For a larger protein with 2000 amino acids, this already becomes:
2000² = 4,000,000 interactions.
And many biologically important proteins are even larger. For example, the well‑known tumor suppressor BRCA2, strongly associated with hereditary breast and ovarian cancer, consists of 3,418 amino acids.
In this case the attention mechanism would need to consider roughly:
3418² ≈ 11.7 million pairwise interactions.
This rapid growth in computation and memory requirements means that longer sequences can quickly exceed GPU memory, often leading to out‑of‑memory (OOM) errors.
If you work with these models yourself, you run into this surprisingly often - something we experienced many times during our own experiments, and it can definitely drive you a bit crazy. Even during inference, models that appear relatively modest in size (for example 7B parameters) can still run into memory limits once sequence lengths become large.
In natural language processing, many tasks can still be solved with relatively small models or shorter context windows, because the most important relationships in text tend to occur locally within sentences or paragraphs.
Genomics is different.
Because biologically meaningful interactions can span extremely long genomic distances, models often need to operate at much larger sequence scales while still preserving nucleotide‑level resolution.
This makes it harder to build smaller, lightweight models that remain biologically useful. As a result, scaling genomic AI often requires substantial computational resources.
Developing new architectures that handle long sequences more efficiently is therefore not only a technical challenge, it is also important for making genomic AI more accessible to researchers with limited compute resources.
And in genomics the pressure is even greater: regions relevant for gene regulation can span hundreds of thousands to millions of base pairs, far beyond what standard transformer architectures can easily handle. This is one of the main reasons researchers are now exploring new model architectures specifically designed for biological sequences, capable of capturing long‑range dependencies without the quadratic scaling of traditional transformers.
🔤 The tokenization challenge
Tokenization (simplified example):
Genomics | is | fascinating
In language models, tokenization means breaking text into smaller pieces called tokens, which can then be converted into numerical vectors and processed by a neural network.
The example above is simplified. In practice, most modern language models use subword tokenization methods that learn frequently occurring character patterns from the training data and turn them into tokens. Rather than splitting strictly by words, these methods produce a vocabulary that efficiently represents common patterns while remaining flexible enough to handle rare ones.
For example, a tokenizer might split a sentence like this:
Gen | omics | is | fasc | inating
DNA sequences, however, do not contain spaces, punctuation, or obvious word boundaries, which makes tokenization less straightforward. Researchers have explored several different approaches.
🔬 1. Nucleotide-level tokenization
The simplest approach is to tokenize letter by letter:
A | T | G | C | C | A | T | G | C
This preserves full sequence resolution but makes it computationally more difficult for models to capture longer biological motifs that span multiple nucleotides.
This full resolution is especially valuable for tasks like variant effect prediction, where a single nucleotide change can be biologically decisive. Because each base maps to exactly one token, that signal is never blurred across neighboring tokens. Models such as Nucleotide Transformer v3 use this single-nucleotide approach.
🪟 2. k-mer tokenization
A sliding window of length k moves across the sequence, producing subsequences.
With an overlapping window, it advances one nucleotide at a time.
Example with k = 3 (overlapping):
ATGCCATGC → ATG | TGC | GCC | CCA | CAT | ATG | TGC
These fragments can help models recognize recurring sequence patterns. This overlapping approach was used in DNABERT.
However, overlapping k-mers introduce an information leakage problem: because neighboring tokens share most of their nucleotides, a masked token can often be reconstructed from its neighbors, making the pretraining task too easy and weakening what the model actually learns.
An alternative is non-overlapping k-mers, where the window jumps ahead by k positions each time.
Example with k = 3 (non-overlapping):
ATGCCATGC → ATG | CCA | TGC
Here nine nucleotides collapse into three tokens. Newer models such as the Carbon family use non-overlapping 6-mer tokenization for exactly this reason: shortening the sequence by a factor of k dramatically cuts the O(n²) cost of attention: reducing the token count 6× shrinks it by roughly 36×.
The Nucleotide Transformer originally relied on non-overlapping k-mer tokenization too, before later migrating to single-nucleotide tokenization alongside other architectural changes - a good example of how the field is still refining these choices.
🧠 3. Learned tokenization (Byte Pair Encoding)
Some models borrow tokenization methods directly from natural language processing, such as Byte Pair Encoding (BPE). Instead of using a fixed window size, BPE learns frequently occurring sequence fragments from the training data and turns them into tokens - building a vocabulary that reflects the actual patterns present in the genome. Models such as DNABERT-2 and GROVER take this approach.
BPE was originally a compression method, and that remains its main advantage here: frequent fragments collapse into single tokens, so long stretches of DNA are represented by far fewer tokens. This shortens the sequence several-fold, expanding how much genome a model can see at once while lowering training cost.
However, BPE also introduces trade-offs. The same stretch of sequence can be split into different tokens depending on where it falls, making the representation inconsistent. And because bases are bundled into larger tokens, the model loses a clear view of the individual nucleotides - a drawback for tasks that depend on single-base precision, exactly where nucleotide-level tokenization is strongest.
The important takeaway is that there is no single standard yet. Different models use different tokenization strategies, and researchers are still actively exploring which representations work best for genomic data.
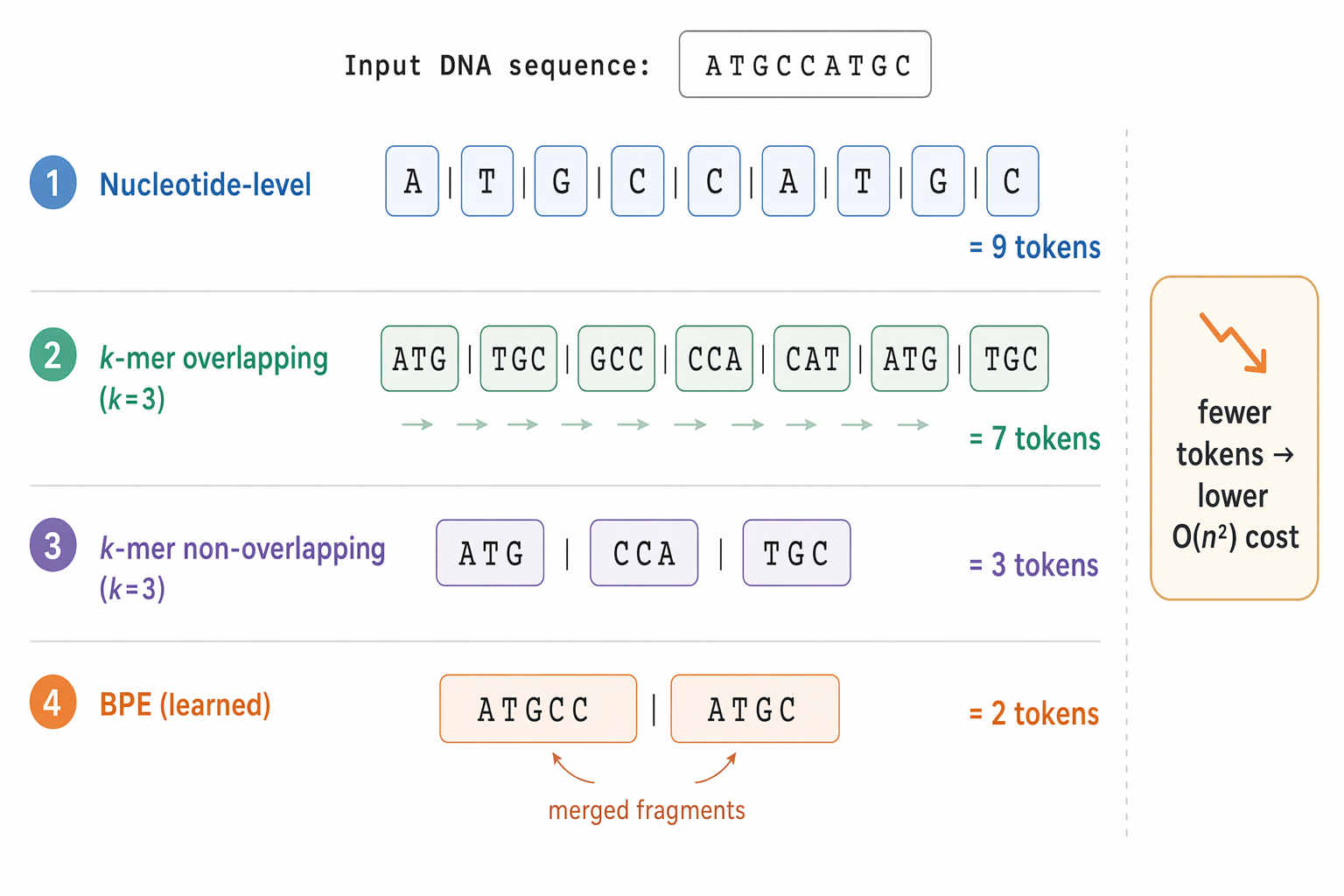
Comparison of four DNA tokenization schemes applied to the sequence ATGCCATGC: nucleotide-level (9 tokens), overlapping k-mer with k=3 (7 tokens), non-overlapping k-mer with k=3 (3 tokens), and learned BPE (2 merged tokens, example split), illustrating that fewer tokens mean lower O(n²) cost
For a deeper dive into genomic tokenization, see: https://academic.oup.com/bioinformatics/article/41/9/btaf456/8237360
🏋️ How genomic language models are trained
Interestingly, even though tokenization strategies differ between natural language and genomic sequences (whether DNA, RNA, or amino acid sequences), the training paradigm is often very similar.
Most genomic foundation models follow the same two-stage recipe that made modern language models successful:
- Pretraining on large amounts of raw, unlabeled sequence, and
- Finetuning on a smaller, labeled dataset for a specific biological task.
The first stage relies on self‑supervised learning. Instead of depending on manually labeled biological data, the model learns directly from raw sequence by predicting parts of it from the surrounding context.
Two main pretraining objectives are borrowed from natural language processing.
Masked training: Masked Language Modeling (MLM)
Parts of the sequence are hidden, and the model learns to predict the missing tokens using context from both directions.
Example:
ATG [MASK] TGC
Because it can look both left and right, the model becomes very good at understanding the meaning and context of sequences. This bidirectional approach powers BERT in natural language and, in genomics, models like DNABERT and the Nucleotide Transformer.
Autoregressive training: Causal Language Modeling (CLM)
Here the model only ever looks left, predicting the next token one step at a time.
Example:
ATG → predict next token
ATGC → predict next token
Because it learns to generate sequence, it can also design new ones which is why GPT-style models such as Evo 2 are particularly exciting for synthetic biology.
![Two-panel comparison of pretraining objectives: Masked Language Modeling (MLM), which is bidirectional and predicts a hidden token (ATG [MASK] TGC) from both directions e.g. Nucleotide Transformer; and Causal Language Modeling (CLM), which is left-to-right and predicts the next token (ATG → ?) from the left only e.g. Evo 2](/images/mlm_clm_image.png)
Two-panel comparison of pretraining objectives: Masked Language Modeling (MLM), which is bidirectional and predicts a hidden token (ATG [MASK] TGC) from both directions e.g. Nucleotide Transformer; and Causal Language Modeling (CLM), which is left-to-right and predicts the next token (ATG → ?) from the left only e.g. Evo 2
Once pretraining is complete, the model is often finetuned: its general sequence representations are adapted, using a comparatively small labeled dataset, to a concrete downstream task such as identifying regulatory elements or predicting the effect of a mutation.
But finetuning is not always necessary. Some models are powerful enough to be used zero-shot so they are applied directly to a task without any task-specific training. Evo 2, for instance, can estimate the effect of a genetic variant by running both the original (wild-type) and the mutated sequence through the model and comparing their internal representations. If the variant meaningfully shifts the model’s hidden states relative to the wild type, that shift can flag a functionally important, potentially disease-relevant, change.
Either way, the model learns patterns, dependencies, and structural signals directly from large amounts of biological sequence data without requiring manual annotations.
But does pretraining actually help?
Recently, this assumption has been challenged. Several studies found that genomic models with randomly initialized weights often performed on par with, or even better than, their pretrained counterparts, and that the learned representations sometimes offered little advantage over simply one-hot encoding the raw sequence. (Sources: https://www.biorxiv.org/content/10.1101/2024.12.18.628606v1.full.pdf+html and https://academic.oup.com/bib/article/27/1/bbag011/8456488)
In my opinion, though, this critique deserves a counter-argument. The fact that a model like Evo 2 can predict variant effects zero-shot, with no task-specific training at all, is hard to square with the claim that pretraining learns nothing useful. If the representations carried no biological signal, there would be nothing for a zero-shot method to exploit in the first place.
From my point of view, a more likely picture is this: pretraining builds broad, general-purpose representations that are useful across many tasks, whereas a randomly initialized model finetuned on a single benchmark simply reshapes its weights to fit that one task. It may match the pretrained model on that specific problem, yet without the same transferability. Pretraining’s advantage also tends to be largest when labeled data is scarce, which is often the case in genomics.
So the honest answer is probably that it depends: on the model, its scale, the training objective, and the task at hand. What these debates really make clear is that being “foundational” for biology, in the way large language models are for text, cannot simply be assumed. It has to be demonstrated.
🚀 What can genomic language models be used for?
Once trained on large amounts of biological sequence data, genomic language models can be applied to a wide range of tasks, many of which are difficult to solve with traditional methods.
Some emerging applications include:
-
De novo sequence generation: generating entirely new DNA or protein sequences with desired properties, which is particularly interesting for synthetic biology and protein design.
-
Zero‑shot variant effect prediction: estimating how genetic mutations might affect gene function without requiring task‑specific training data.
-
Identification of new genomic elements: discovering previously unknown promoters, enhancers, or other functional regions directly from sequence patterns.
-
Mapping regulatory relationships: learning how different genomic regions influence gene expression and regulatory networks.
-
Predicting 3D genome organization: inferring chromatin interactions and structural genome features from sequence information.
-
Understanding non‑coding regions: analyzing the vast parts of the genome that do not encode proteins but still play important regulatory roles.
-
Reconstructing missing sequences: filling gaps in genome assemblies or predicting plausible sequences in poorly characterized organisms.
-
Learning biological embeddings: creating numerical representations of sequences that can be used for downstream tasks such as clustering, similarity search, or retrieval‑augmented analysis.
These capabilities are still evolving, but they illustrate why genomic language models are attracting growing interest across computational biology, medicine, and biotechnology.
🏗️ New architectures for genomic AI
Some models still rely on classical transformer architectures applied directly to genomic sequences.
Examples include:
- DNABERT
- GROVER
- Carbon models
But as researchers began pushing toward larger genomic contexts, new architectures started to emerge.
One interesting example is Evo 2, which is based on a Hyena-based architecture (StripedHyena). (Source: https://www.nature.com/articles/s41586-026-10176-5)
Instead of relying purely on quadratic attention between all tokens, Hyena models use long convolutional operators that capture relationships across very long sequences while scaling much more efficiently.
This allows models like Evo 2 to process genomic contexts that would be extremely difficult to handle with standard transformer attention.
Training models at this scale still requires enormous compute resources. Evo 2‑style models, for example, are typically trained on modern GPU clusters such as NVIDIA H100 systems.
Another fascinating direction is represented by AlphaGenome. (Source: https://www.nature.com/articles/s41586-025-10014-0)
Instead of changing the attention mechanism itself, AlphaGenome focuses on distributed sequence modeling at scale.
During training, genomic sequences are broken into chunks that are distributed across large TPU clusters, while the architecture still allows nucleotides across different chunks to attend to each other.
This makes it possible to model very long genomic regions without fitting the entire sequence onto a single accelerator.
Interestingly, AlphaGenome also revives the idea of a much older architecture: the U‑Net, originally introduced back in 2015 for biomedical image segmentation. (Source: https://arxiv.org/abs/1505.04597) A U‑Net first compresses its input step by step into a coarse, high‑level summary (the “encoder”), then expands it back to full resolution (the “decoder”), using skip connections to carry fine‑grained detail across the two halves. Applied to genomics, this lets AlphaGenome combine a broad view of long‑range context with nucleotide‑level resolution, a nice reminder that progress in the field often comes from creatively reusing older ideas, not only from inventing entirely new ones.
From an engineering perspective, this is a remarkable achievement: it effectively spreads the genome across many devices while maintaining global interactions between sequence elements.
AlphaGenome is also interesting from a biological perspective. Through its training objective, the model learns signals that are closely related to epigenetic regulation and chromatin structure, allowing it to implicitly capture aspects of genome regulation beyond the raw sequence itself.
These examples illustrate an important trend in the field.
Understanding genomes with AI is not only about scaling model size, it also requires new architectures and clever engineering strategies to deal with the unique structure of biological sequences.
🌐 The bigger picture: biology is multimodal
Even the DNA sequence alone does not tell the whole story.
The genome is the raw DNA sequence, but biological information is spread across many more layers. How the sequence is chemically modified (epigenetics), which genes are switched on (RNA), the proteins they build, and how everything folds into 3D structures all shape what actually happens in a cell. Measuring and combining these layers is the domain of multi‑omics.
DNA → RNA → proteins → 3D structures → cellular and tissue specific regulation → epigenetics
Future AI models will likely need to integrate all of these signals.
In other words, the next generation of models may not just be genomic language models, but multimodal biological foundation models. For a great read pointing in exactly this direction, toward generalist biological AI spanning DNA, RNA, proteins, and cellular systems, see: Generalist biological artificial intelligence in modeling the language of life
The idea that DNA could be treated like a language sparked an entire field of research.
But the deeper scientists explore this analogy, the clearer it becomes:
The language of life follows very different rules than human language.
Understanding those rules is now driving the development of entirely new AI architectures, designed not for text, but for biology.
And that could ultimately transform how we understand disease, develop therapies, and design biological systems.